What we do
Complex traits (e.g. yield, resistance to biotic and abiotic stresses, nutrition efficiency, etc.) are vital to crop improvement toward productive and sustainable agriculture. Given the importance of wheat in the food system, Lu Lab uses interdisciplinary approaches, combining genomics, quantitative genetics, population genetics, evolution, and computer science, to dissect complex traits of wheat. Primary research questions we are trying to address include, but are not limited to.
1) What are the genetic architectures of complex traits?
2) What are the causative variants for phenotypic variation?
3) Why do individual plants respond differently to the same environmental change?
4) What determines the evolvability or adaptive potential of phenotypes and species?
5) How to apply the insights obtained from 1-4 and design better crops?
By answering these questions, we hope to improve genomic prediction/selection and develop a new system of quantitative trait breeding using technologies of genome editing and synthetic biology.
Challenges
Quantitative traits are important for crop improvement, but difficult to work with, because 1) hundreds to thousands of loci are controlling the traits; 2) each locus has small effect. The challenge is to identify thousands of loci and accurately estimate their effects together, which generally requires a large sample size in GWAS. However, due to the relatively higher cost for collecting phenotypes (compared with human genetic studies), it is not feasible to build a population of 1 million plants to estimate these small effects. There has to be another route to dissect complex trait, which needs to be creative, efficient, and cost-effective.
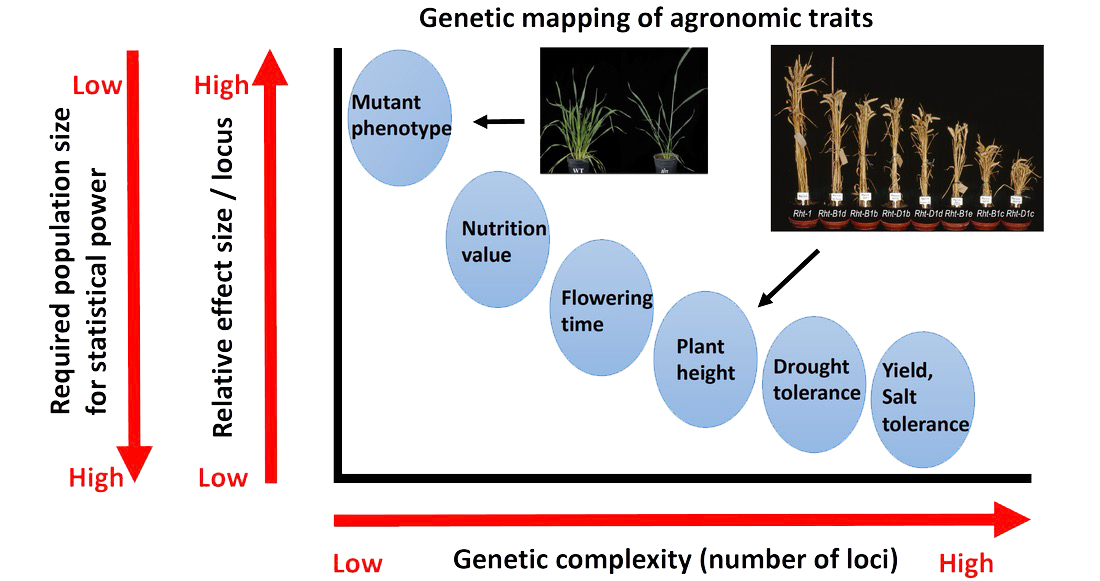
Our Solution
We use functional genome prediction approach to pinpoint trait controlling variants and estimate their effect. By using evolution (nature’s experiment one billion years), decomposing complex traits to molecular level, combining with field trials and high-throughput sequencing, utilizing the power of optimization and machine learning approaches, we predict causal variants underneath important agronomic traits. The successful development of functional genome prediction approach will tremendously expand the application of genomic editing technologies in crops.